基于SVM的遗传疾病的致病位点预测分析
小组成员
- 蔡鑫奇:2620160006
- 牟宇超:2620160005
- 武 阳:2620160010
1. 问题背景
人类健康问题是当今社会中备受关注的热点。人类疾病,特别是多基因遗传病相关基因的定位,是目前生物医学研究中十分重要的一项。大量研究表明,人类的性状差异与对药物和疾病的易感性都可能与基因某些位点相关。因此,定位与性状或疾病相关联的基因中的位点,有利于生物医学中对一些疾病遗传机理的研究,并方便对相应疾病的治疗和预防。
2. 问题描述
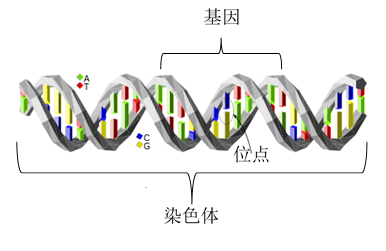
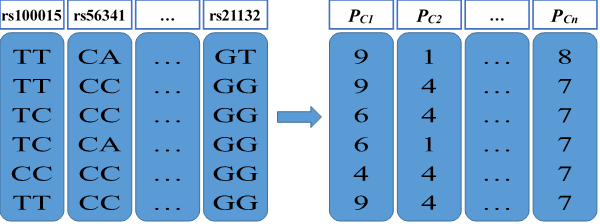
近年来,随着计算机应用技术的发展以及大数据时代的到来,人们对人体基因与遗传性疾病的研究越来越深入了。研究人员大都采用全基因组的方法来确定致病位点或致病基因,具体做法是:招募大量志愿者(样本),包括具有某种遗传病的人和健康的人,通常用1表示病人,0表示健康者。对每个样本,采用碱基(A,T,C,G)的编码方式来获取每个位点(在组成DNA的数量浩瀚的碱基对中,有一些特定位置的单个核苷酸经常发生变异引起DNA的多态性,我们称之为位点,染色体、基因和位点的结构关系见图1.)的信息(因为染色体具有双螺旋结构,所以用两个碱基的组合表示一个位点的信息);如表1中,在位点rs100015位置,不同样本的编码都是T和C的组合,有三种不同编码方式TT,TC和CC。研究人员可以通过对样本的健康状况和位点编码的对比分析来确定致病位点,从而发现遗传病或性状的遗传致病机理。
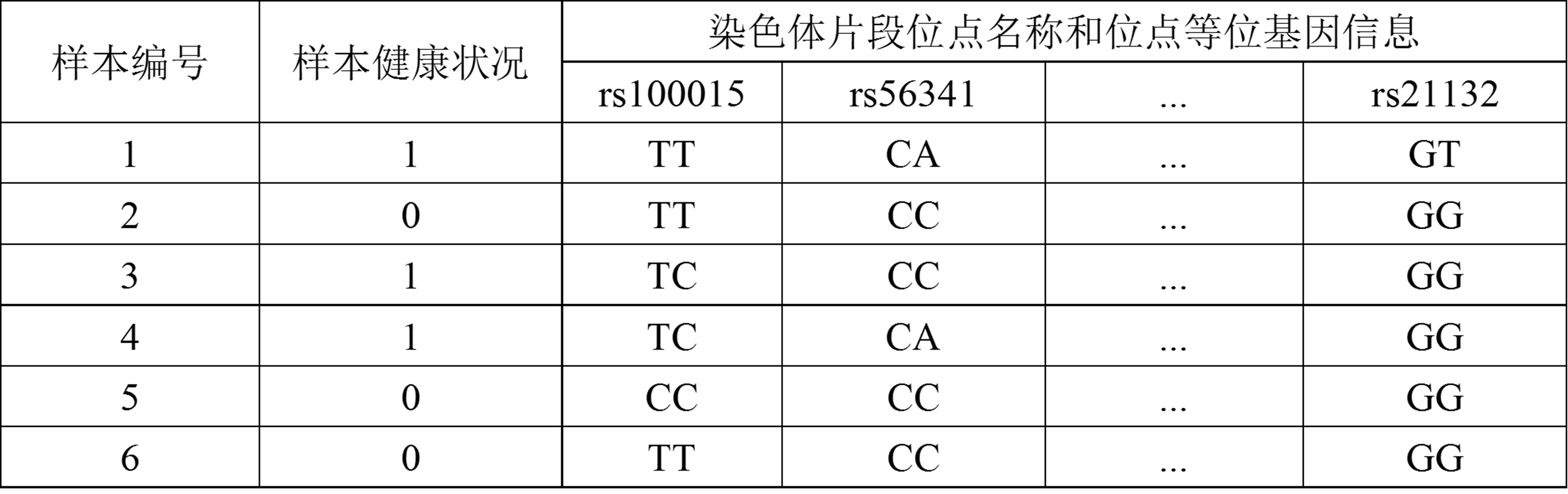
注:以6个样本为例,其中3个病人,3个健康者,位点名称通常以rs开头。
我们要研究的主要问题是:分析1000个样本在某条有可能致病的染色体片段上的9445个位点的编码信息和样本患有遗传疾病A的信息。对致病位点进行检测,预测某种疾病的致病位点,其实就是判断不同位点对该疾病的影响程度,即判别每个位点的属性关于致病与非致病类别的分类精度。
3. 数据模型
数据集(gene_pheno_dataset)大小:27.1 MB
包含文件:

数据清洗及分类提取后的数据集如下:
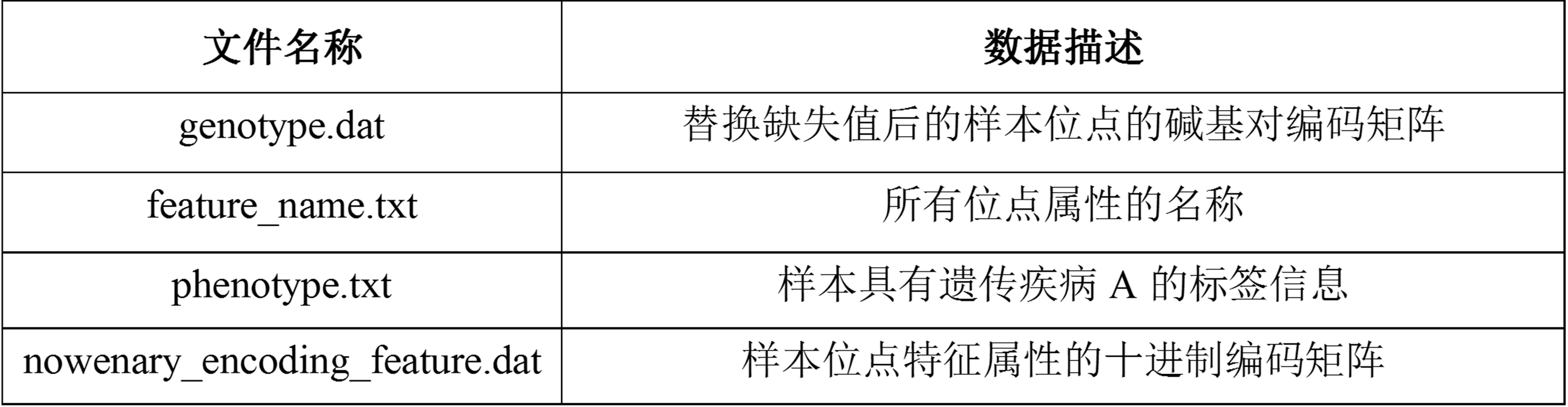
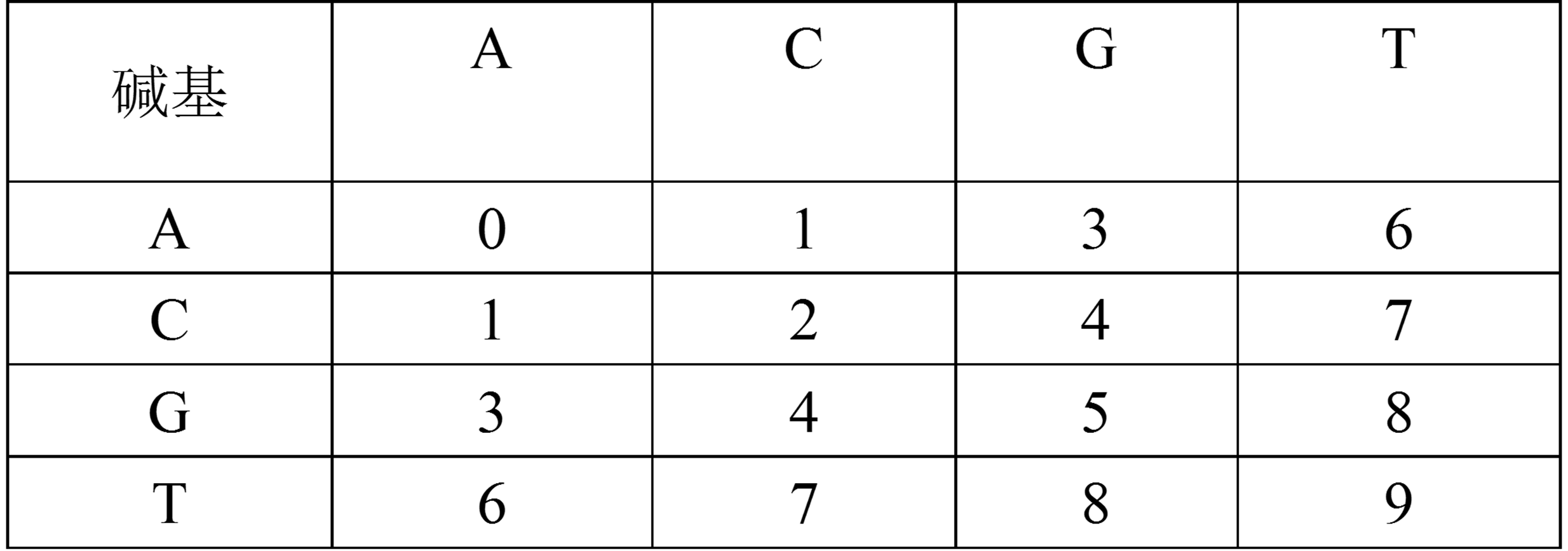
我们使用了位点测试数据集,来自1000个可能致病的染色体片段试验检测结果,标签分布为500个无病染色体使用0表示,500个患病染色体使用1表示,且每个致病染色体上有9445个碱基对,以此作为位点。采用十进制{0,1,2,3…}编码将每个碱基对转化成数据编码方式,以便于数据分析。“AA”为“0”;“AC”为“1”;“AG”为2;“AT”为3…“TT”为9,详见碱基对编码表2(其中{AC,CA};{CG,GC};…碱基对表示方式相同)。
另外,位点中出现字符‘I’和‘D’,根据说明,分别用‘T’和‘C’代替。 由于所有样本序列上的本一个二核苷酸位点代表了一个属性,本文总共有9445个位点即9445个不同的属性,这些属性由十进制表示(见图2.)。其中,属性列中PC1~PCn 表示9445个不同的属性指标;AA,AC,AG,AT,…,TT表示16中不同的原始二核苷酸。
4. 算法设计原理与实现
本文要解决的是寻找遗传疾病A的致病基因位点,通过之前的分析,也就是对9445个位点属性进行分类预测,一类是致病基因位点(标签为label=1),另一类是非致病基因位点(标签为label=0),因而这个问题就转化为一个分类预测问题了。 算法设计上,我们分为两大块,包括:主函数(main.m)设计、预测函数(predictFunc_svm.m)设计。
4.1 主函数设计模块
基于本文要解决的问题,算法主函数要得到的结果应包括:9445个位点属性的预测精度(输出predict_accuracy.txt)、预测精度的降序排列结果及对应的位点序号(输出predict_accuracy_desc.txt)、Top n 预测精度及对应预测精度所在的位点名称(n取10)。 主函数算法实现步骤:
1) 开始; 2) 加载十进制编码9445个位点的所有属性数据all_feature(i,j); 3) 数据属性归一化处理,将十进制数据变换到(0, 1)区间上; 4) 循环每个位点,调用预测函数对每列属性进行该疾病的预测,得到预测精度accuracy; 5) 对预测结果降序排列,即预测精度 accuracy 从高到低排列; 6) 选出Top n 预测精度及对应预测精度所在的位点; 7) 结束。
其中,数据归一化处理过程中,对于第i个样本的第j个位点编码all_feature(i,j)做如下变换得到归一化的属性编码数据:
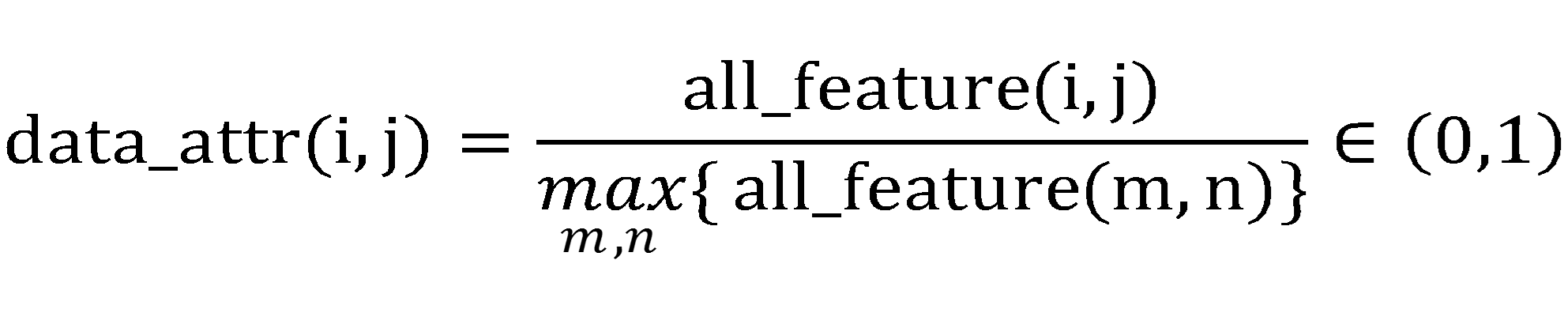
4.2 预测函数设计模块
本文采用K折交叉验证的实验测试方法及支持向量机(SVM)分类器来构造位点属性的分类预测函数,用于获取预测精度。这一部分目前仍处于完善之中,预测函数的程序仍在调试之中。
5. 后续工作
1) 继续调试程序,对预测函数(predictFunc_svm.m)进行完善; 2) 利用实验数据训练出预测分类模型,并通过该分类模型预测出该疾病最可能的几个致病位点; 3) 利用MATLAB运行出实验结果,并完成最终报告。
注:数据清洗及分类提取后的数据集:文件的github仓库 (WashedDataset文件夹下)
- genotype.dat
- feature_name.txt
- phenotype.txt
- nowenary_encoding_feature.dat